Heterozygous β-globin gene mutation causing reduced β-chain production. Results in a mild, asymptomatic microcytic anemia often discovered incidentally.
It is benign for the patient but important to recognize to avoid misdiagnosis as iron deficiency and unnecessary iron therapy. Also, two carriers can have a child with severe thalassemia major, so identifying carriers allows for genetic counseling and prenatal diagnosis.
Usually asymptomatic. May have mild fatigue or no symptoms at all. Often found on routine blood work showing low MCV (~65–75 fL) and mild anemia (Hb ~10–13).
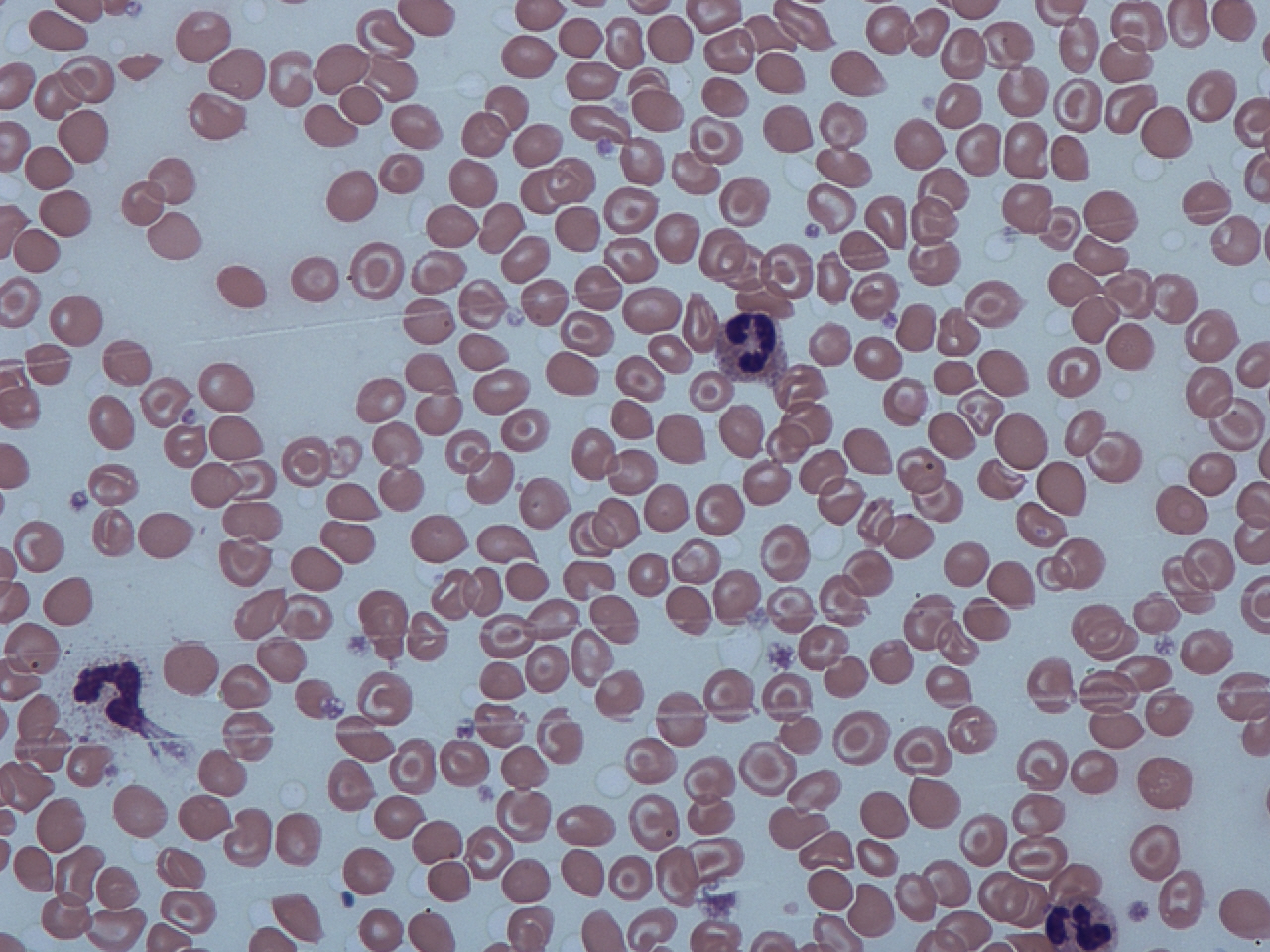
Labs: Iron studies normal. RBC count normal or slightly high (reflecting many small RBCs). Smear: target cells, mild anisocytosis. Hemoglobin electrophoresis: ↑HbA2 (>3.5%) is the hallmark; HbF may be slightly elevated.
Often noted in context of a patient not responding to iron supplements or during screening in pregnancy. Ethnic background (Mediterranean, Asian, African) can raise suspicion.
Differentiate from iron deficiency: β-thal trait has normal ferritin and often a high RBC count (iron deficiency: low ferritin, low RBC count). Mentzer index <13 suggests thalassemia.
Confirm diagnosis with electrophoresis (run when iron replete): elevated HbA2 confirms β-thal minor. Note that concurrent iron deficiency can mask this (normalize iron first).
Once diagnosed, avoid unnecessary treatments. Instead, provide patient education and offer partner testing if of childbearing age due to reproductive implications.
Condition
Distinguishing Feature
Iron deficiency anemia (mild)
more common; usually low ferritin, low RBC count, responds to iron therapy
Alpha-thalassemia trait
also mild microcytosis; electrophoresis is normal (no elevated HbA2)
Anemia of chronic disease
microcytic in chronic inflammation; features of underlying disease present (e.g., arthritis, chronic infection)
No specific treatment is required for the mild anemia. Avoid iron supplementation unless iron deficiency is also present (to prevent iron overload).
Advise folic acid supplementation if there's any ongoing hemolysis (usually not significant in trait).
Genetic counseling: Explain autosomalrecessive inheritance. If both partners have trait, discuss prenatal diagnosis options (CVS/amniocentesis) to detect severe thalassemia in fetus.
HbA2 >3.5% on electrophoresis = β-thalassemia trait.
β-thalassemia trait is often misdiagnosed as iron deficiency – check iron studies before giving iron.
Carriers often have relatives with similar mild anemia; a family history of "low blood count" can be a clue.
Do not overlook a β-thalassemia minor in a pregnant patient – unnecessary iron can accumulate and fail to correct anemia; ensure proper diagnosis.
If a trait carrier's child has unexplained anemia early in life, test for more severe thalassemia (child could have inherited severe form if the other parent is also a carrier).
Microcytosis on CBC but normal iron → suspect thalassemia trait.
Perform hemoglobin electrophoresis (after confirming no iron deficiency): if ↑HbA2 is seen → β-thalassemia trait confirmed.
Reassure patient; no active treatment needed except avoiding inappropriate iron. Document carrier status in medical record.
Test the partner if planning children; refer to genetic counseling if both are carriers.
Young adult with persistent microcytosis despite adequate iron, RBC count normal, and elevated HbA2 on electrophoresis → β-thalassemia minor.
Prenatal screening reveals both parents have increased HbA2 levels → both are β-thalassemia carriers (risk of Cooley's anemia in offspring).
Case 1
A 25-year-old woman is told she has iron deficiency due to a microcytic anemia, but iron supplements haven't raised her hemoglobin.
Case 2
During premarital testing, a man is found to have microcytosis with elevated HbA2. His fiancée's labs show the same abnormality.
Blood smear with target cells (thin, bullseye-appearing RBCs). Target cells are often seen in beta-thalassemia minor.