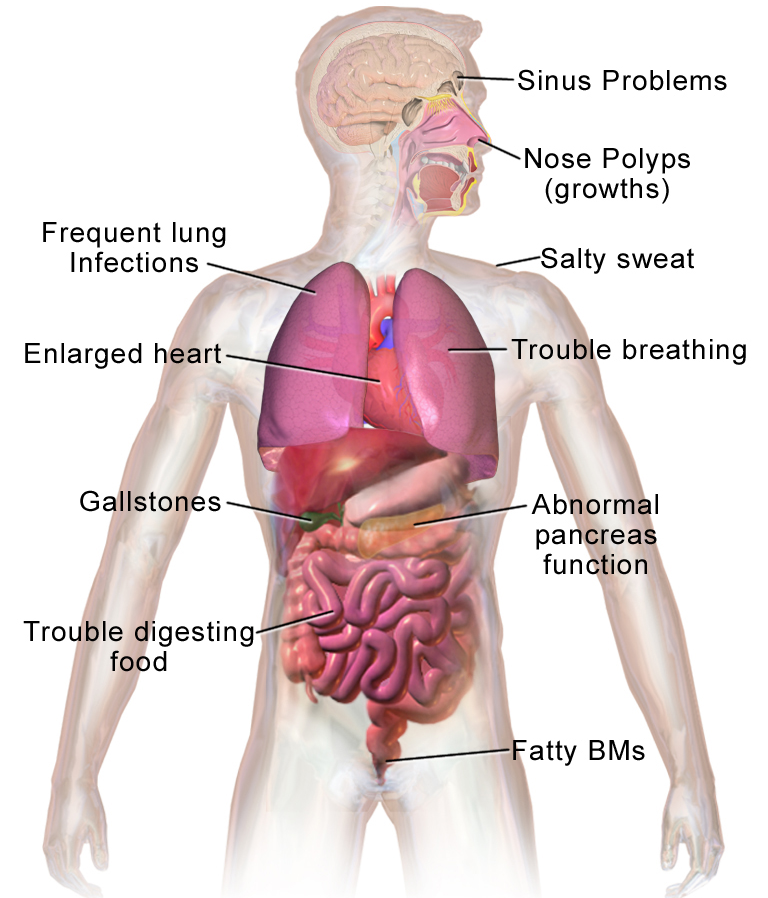
Autosomalrecessive disorder of CFTR (chloride channel) causing abnormally thick secretions in exocrine tissues and multi‑organ damage (especially lungs and pancreas).
Most common lethal genetic disease in Caucasians (≈1:3500 births). Once fatal in childhood, now many survive into middle age due to new therapies (CFTR modulators). A classic exam disease (e.g., salty sweat, Pseudomonas infections, meconium ileus).
Neonate: meconium ileus causing neonatal intestinal obstruction (no meconium in 48 hrs, bilious vomiting) → strongly suggests CF.
Infancy/childhood: failure to thrive (poor growth) with foul-smelling, greasy stools (pancreatic insufficiency) and recurrent respiratory infections (cough, pneumonia). Typical organisms: Staph aureus in infancy, then Pseudomonas aeruginosa in early childhood; digital clubbing often develops.
Adolescence/adulthood: chronic productive cough, nasal polyps, and bronchiectasis on imaging. Almost all males have infertility (absence of vas deferens), and many patients develop CF‑related diabetes by young adulthood.
Suspect CF in any infant with meconium ileus or failure to thrive with steatorrhea, or in young adults with bronchiectasis and unexplained infertility.
Diagnostic gold standard: sweat chloride test (pilocarpine iontophoresis); sweat [Cl–] ≥60 mmol/L on two occasions is diagnostic. If sweat test is borderline, perform expanded CFTR genetic testing.
After diagnosis, genotype analysis of the CFTR gene guides therapy (eligibility for CFTR modulators). Also assess organ involvement: perform baseline spirometry, sputum cultures (often showing Pseudomonas), and evaluate pancreatic function (fecal elastase).
Distinguish CF from primary ciliary dyskinesia: CF has positive sweat test and pancreatic insufficiency, whereas ciliary disorders have normal sweat chloride and often situs inversus.
neonatal distal bowel obstruction due to colonic aganglionosis; no pancreatic insufficiency or respiratory disease
CFTR modulator drugs (targeted to mutation): e.g., ivacaftor for G551D gating mutation, or combination correctors/potentiators (elexacaftor/tezacaftor/ivacaftor) for ΔF508; these markedly improve lung function.
Aggressive pulmonary care: airway clearance via chest physiotherapy (percussion vest) and inhaled mucolytics (e.g., dornase alfa, hypertonic saline); bronchodilators to improve airflow; and frequent antibiotics to treat/prevent infections (including inhaled tobramycin for Pseudomonas).
Nutritional support: high-calorie, high-salt diet with pancreatic enzyme replacement and fat-soluble vitamin supplements to ensure growth. Screen for and manage complications (e.g., insulin for CF-related diabetes, ursodeoxycholic acid for liver disease).
Advanced disease: consider lung transplantation for end-stage lung failure (improves survival when FEV₁ <30% or if frequent exacerbations, pneumothorax, or massive hemoptysis).
Parents kissing their baby and noticing a salty taste (salty skin) is a classic clue (high sweat chloride).
Meconium ileus in a newborn is CF until proven otherwise.
Acute worsening of breathing in CF → consider spontaneous pneumothorax (due to apical blebs); requires prompt chest tube and management.
Newborn screening (↑ immunoreactive trypsinogen) positive or suggestive symptoms → evaluate for CF.
Confirm diagnosis: perform sweat chloride test (≥60 mmol/L is diagnostic); if sweat test is indeterminate, do CFTR genetic mutation panel.
If CF confirmed, establish baseline: pulmonary function tests, sputum culture for pathogens, pancreatic function tests (e.g., fecal elastase), and nutritional status assessment.
Initiate comprehensive management early: airway clearance therapy, pancreatic enzymes and nutritional support, and infection prophylaxis/treatment as needed (multidisciplinary CF care).
Follow up regularly at a CF center; monitor lung function and growth, and screen annually for complications (e.g., glucose tolerance test for CF-related diabetes).
Newborn with abdominal distension, no meconium in 48 hrs (meconium ileus) and bilious vomiting → cystic fibrosis.
Toddler with recurrent lung infections (including Pseudomonas), failure to thrive, and foul greasy stools → cystic fibrosis.
Young man with chronic sinus infections, bronchiectasis, and infertility (absent vas deferens) → nonclassic cystic fibrosis.
Case 1
A 2‑year‑old boy is evaluated for chronic cough, frequent diarrhea, and poor weight gain.
Diagram of cystic fibrosis effects on multiple organs (sinuses, lungs, pancreas, intestines), highlighting thick mucus and associated symptoms.