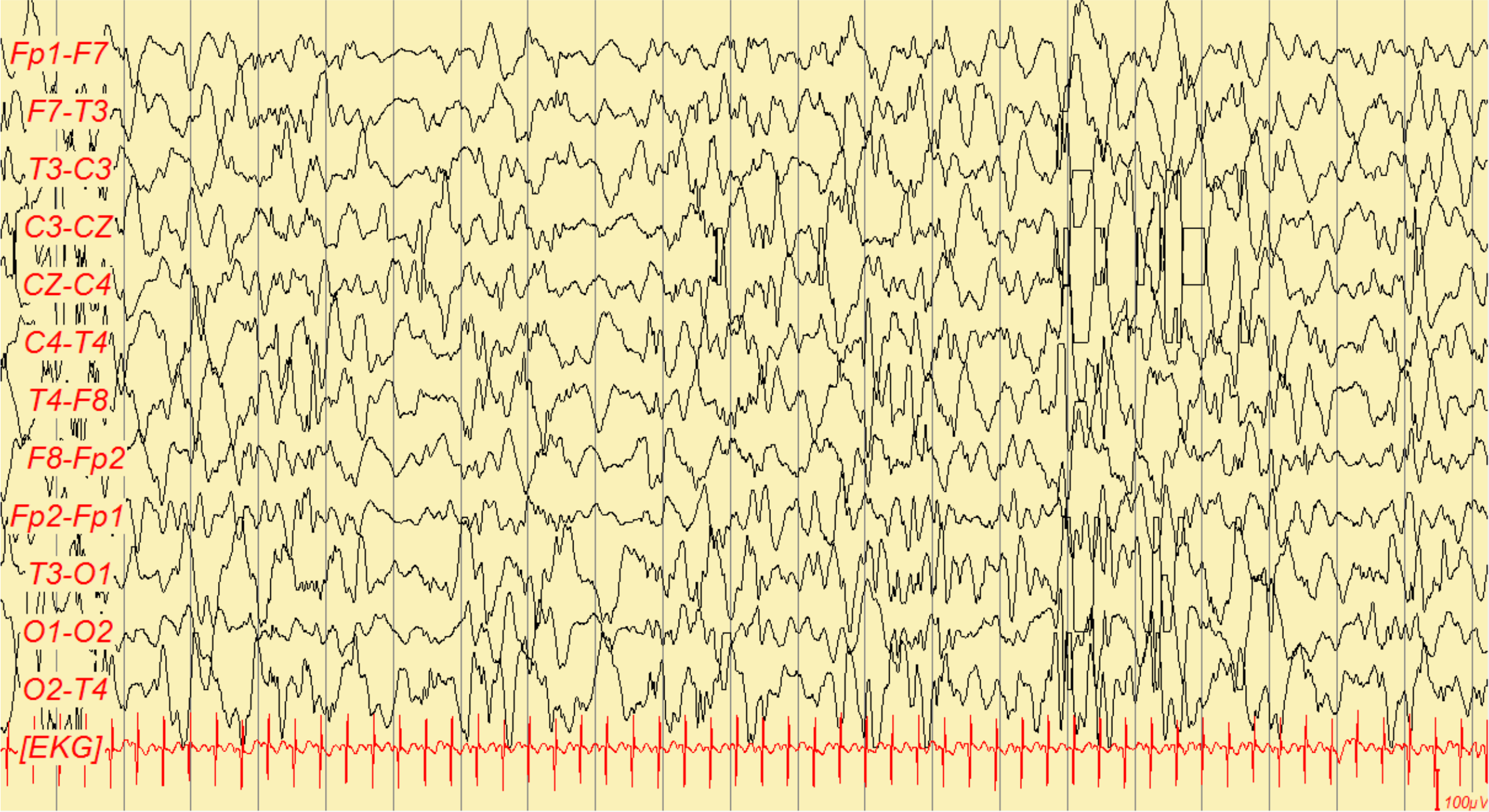
Age-specific epilepsy of infancy defined by a triad: clusters of infantile spasms, an abnormal interictal EEG pattern called hypsarrhythmia, and developmental stagnation or regression.
Considered a catastrophic epilepsy of infancy that can cause permanent developmental loss if not promptly treated. Early recognition (often by its classic triad and associations like tuberous sclerosis) is crucial for improving outcomes.
Typically onset at 3–8 months of age with clusters of sudden, brief symmetric spasms (flexion or extension of the neck, trunk, and limbs) that often occur upon awakening.
Spasms may be subtle early on (e.g., head drops or eye deviation) but often progress to "jackknife" or salaamseizures (infant bows forward at the waist). Clusters can number dozens of spasms, and infants often cry or become irritable afterward.
Developmental skills halt or regress around the time spasms begin (losing milestones or failing to progress). In some cases, a prior abnormality (e.g., brain injury or genetic condition) caused developmental delay even before seizure onset.
Look for underlying causes: tuberous sclerosis (infantile spasms occur in ~50–70% of TSC patients, often with ash-leaf skin spots and other findings), Down syndrome (trisomy 21 increases infantile spasm risk), perinatal brain injury (hypoxic-ischemic encephalopathy, stroke), metabolic disorders, or other CNS malformations. However, ~10–40% of cases are cryptogenic (no identified cause).
If infantile spasms are suspected, obtain an urgent EEG. The hallmark is hypsarrhythmia – a chaotic high-voltage background with multifocal spikes between spasms.
Evaluate for etiologies: perform brain MRI to detect structural lesions (cortical dysplasia, tubers in TSC, etc.), and consider metabolic/genetic testing (e.g., urine amino acids, genetic panels) if initial workup is unrevealing.
Start treatment immediately once West syndrome is diagnosed – do not wait for exhaustive etiologic workup. First-line therapy is typically hormonal: ACTH injections or high-dose oral corticosteroids (e.g., prednisolone).
If tuberous sclerosis is the cause, initiate vigabatrin as first-line, as it often effectively controls spasms in TSC-associated West syndrome.
Monitor clinical response and repeat EEG after treatment initiation to confirm resolution of hypsarrhythmia. If spasms persist, escalate therapy (combine ACTH/steroid with vigabatrin, or try other antiseizure medications) and consider a ketogenic diet or epilepsy surgery for refractory cases.
Condition
Distinguishing Feature
Normal infantile movements
e.g., exaggerated startle (Moro reflex) or shuddering attacks – no EEG abnormalities and infant remains developmentally normal
Sandifer syndrome
GERD-related dystonic posturing (arching of back and neck) in infants, often during/after feeding
Benign myoclonic epilepsy of infancy
brief myoclonic jerks in infants without developmental regression; EEG is not hypsarrhythmic
ACTH (adrenocorticotropic hormone) intramuscular injections are a first-line therapy to suppress infantile spasms (particularly for non-tuberous sclerosis cases). High-dose corticosteroids (like oral prednisolone) are an alternative first-line option.
Vigabatrin (a GABA-transaminase inhibitor) is first-line for West syndrome caused by tuberous sclerosis, and can be used in other cases if ACTH/steroids are contraindicated or ineffective. Monitor for visual field loss with vigabatrin.
Refractory cases: consider adding or switching to other antiseizure meds (e.g., topiramate, valproate), initiating a ketogenic diet (often effective for infantile spasms), or surgical resection if a focal lesion is triggering spasms. Close EEG and developmental follow-up is required.
Older term "salaam seizures" refers to infantile spasms – the infant appears to do quick bowing motions (as in a salaam).
Up to half of infants with West syndrome later develop Lennox–Gastaut syndrome (a severe childhood-onset epilepsy), especially if an underlying brain injury or malformation is present.
Clusters of spasms in an infant (with or without developmental regression) = neurologic emergency. Do not attribute these episodes to benign causes (colic, reflux, startle); urgent evaluation (EEG, neuroimaging) and treatment can prevent further brain injury.
Delayed treatment of infantile spasms is associated with worse developmental outcomes – if West syndrome is even suspected, involve pediatric neurology and start therapy promptly.
Infant <1 year with recurrent brief bilateral spasms ± developmental regression → suspect West syndrome.
Obtain urgent EEG; if hypsarrhythmia is present, this confirms the diagnosis of infantile spasms (West syndrome).
Initiate first-line treatment immediately: ACTH (or high-dose steroid) as soon as possible; use vigabatrin if tuberous sclerosis is identified.
Perform workup for underlying cause in parallel: brain MRI, metabolic and genetic testing to identify any syndromic, structural, or metabolic etiology.
Reassess frequently: track spasm frequency and perform follow-up EEG after treatment to ensure hypsarrhythmia has resolved. If spasms continue, escalate therapy (combine treatments, ketogenic diet, consider surgical evaluation).
A 6-month-old infant who was developing normally now has clusters of brief flexion spasms of the neck, trunk, and arms upon awakening. The baby has lost previously acquired milestones and an EEG shows a chaotic high-amplitude pattern → West syndrome (infantile spasms).
An infant with tuberous sclerosis (hypopigmented "ash-leaf" skin macules and cardiac rhabdomyomas) develops infantile spasms. EEG reveals hypsarrhythmia. The appropriate first-line treatment is vigabatrin in this scenario (West syndrome due to TSC).
Case 1
A 5-month-old girl who previously cooed and smiled now has begun having strange episodes shortly after waking. She briefly jerks her head and arms forward, sometimes in clusters of 5–10, and then cries. Over the past few weeks she has stopped rolling over or babbling as she used to. Exam reveals several hypopigmented "ash-leaf" spots on her trunk.
EEG showing hypsarrhythmia – disorganized high-voltage slow waves and multifocal spikes, characteristic of West syndrome.